Research Directions
Specialization Keywords: atomistic simulation, density functional theory (DFT), solid-state physics, statistical mechanics
-
Battery materials
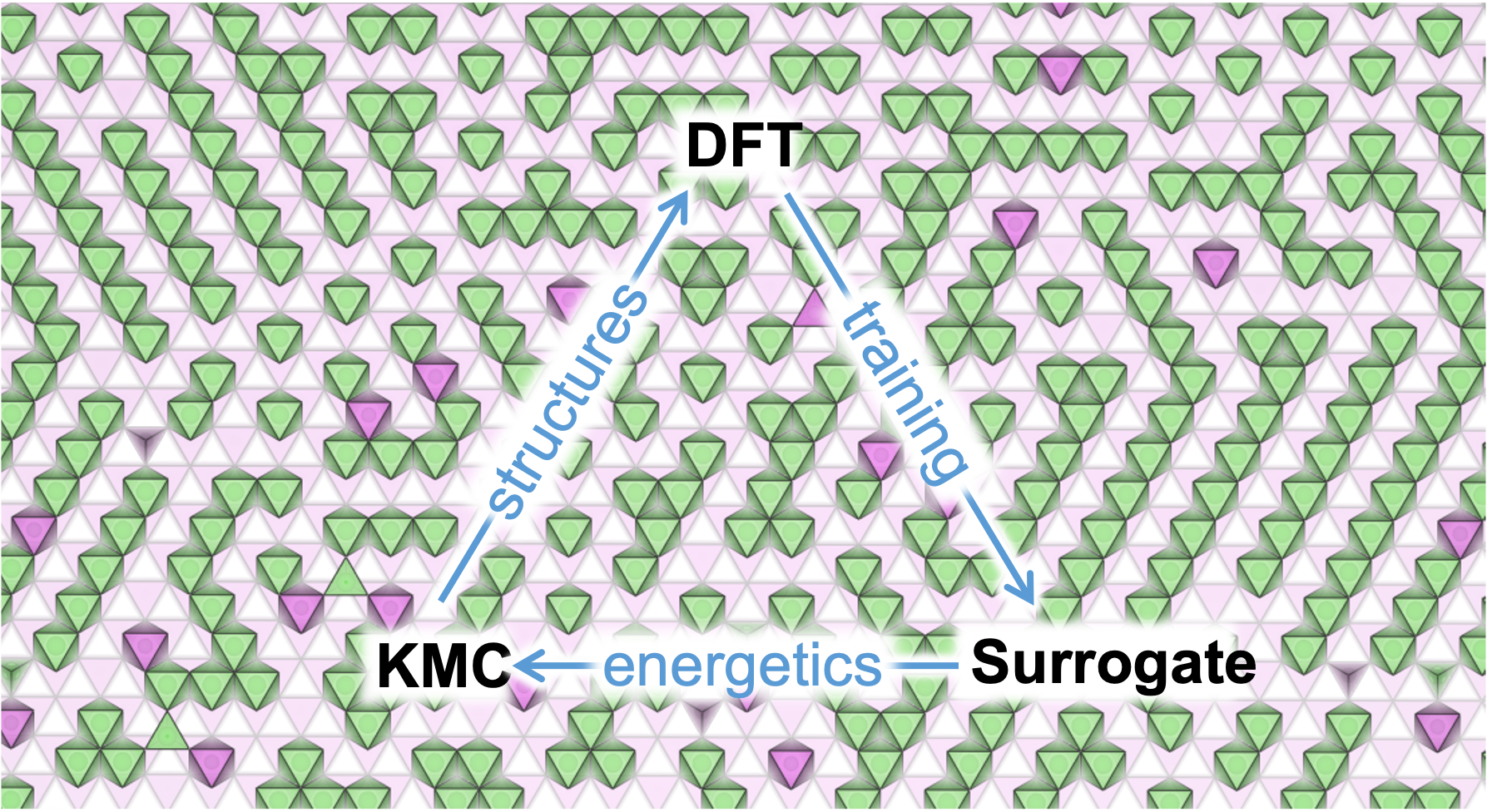
We simulate the motions of intercalating ions (Li+, Na+ etc.) in inhomogeneous local environments, as well as the dynamic rearrangement of host materials. This combination allows us cover three aspects of battery materials from first-principles calculations: synthesis, performance degradation, and characterization.
The computational framework under active development is shown in the schematic. Energies from the density functional theory (DFT) lay the foundation, from which effective interactions between moving ions can be extracted. Then diffusion barriers are acquired with a surrogate model and inputted to kinetic Monte Carlo for long time scale dynamics. Finally, macroscopic characterization signals are generated from the statistical average to make our predictions provable in experiments. We will focus on electrochemical measurements which sensitively reflect the structure-kinetic relationship and are vastly accessible experimentally. These include cyclic voltammetry (CV), potentiostatic intermittent titration (PITT), EIS, galvanostatic intermittent titration (GITT), and Atlung method for intercalant diffusion (AMID).
Currently we are interested in the kinetics of LiFe1-xMxPO4 and LiNi1-xMxO2 where M=Mn, Li.
-
Electrocatalysis
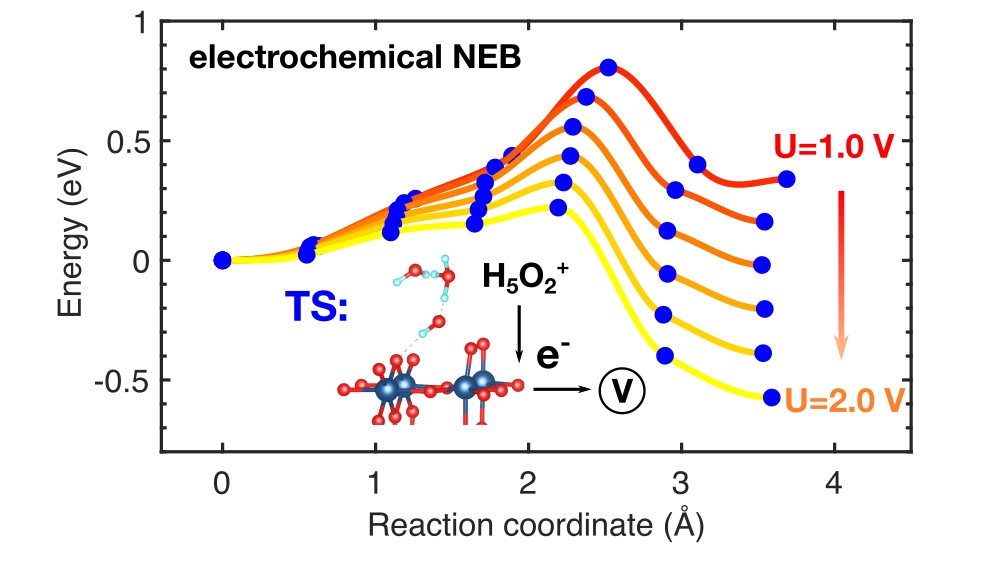
We have developed a set of computational methods to exactly assess the effect of applied voltage on surface reaction rates. The number of electrons is treated as an additional degree of freedom that responses to the electrode potential, so that the electrochemical driving force is directly applied in DFT calculations. This approach can handle partial charge transfer, quantify the symmetry factor of charge transfer, reveal the coupling between net charge and geometry relaxation. We are interested in revisiting the reaction mechanisms in some important electrochemical systems, such as CO2 reduction, OER for water splitting and Zn-O2 battery.
-
Corrosion
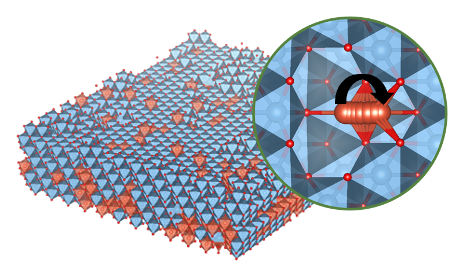
Corrosion and passivation involve cation diffusion, oxygen reduction/evolution and cation dissolution. The computational approaches in both battery and electrocatalysis can be applied here. We are interested in the corrosion of structural alloys, as well as the degradation of batteries and catalysts.
-
Method development
We develop numerical algorithms for long time dynamics, including saddle point search algorithms for off-lattice models and density function theory (DFT) informed kinetic Monte Carlo methods for lattice models. We are also interested in using machine learning to accelerate the above from existing data.
